1BRIUMVI®. Fachinformation. Neuraxpharm, 04/2025
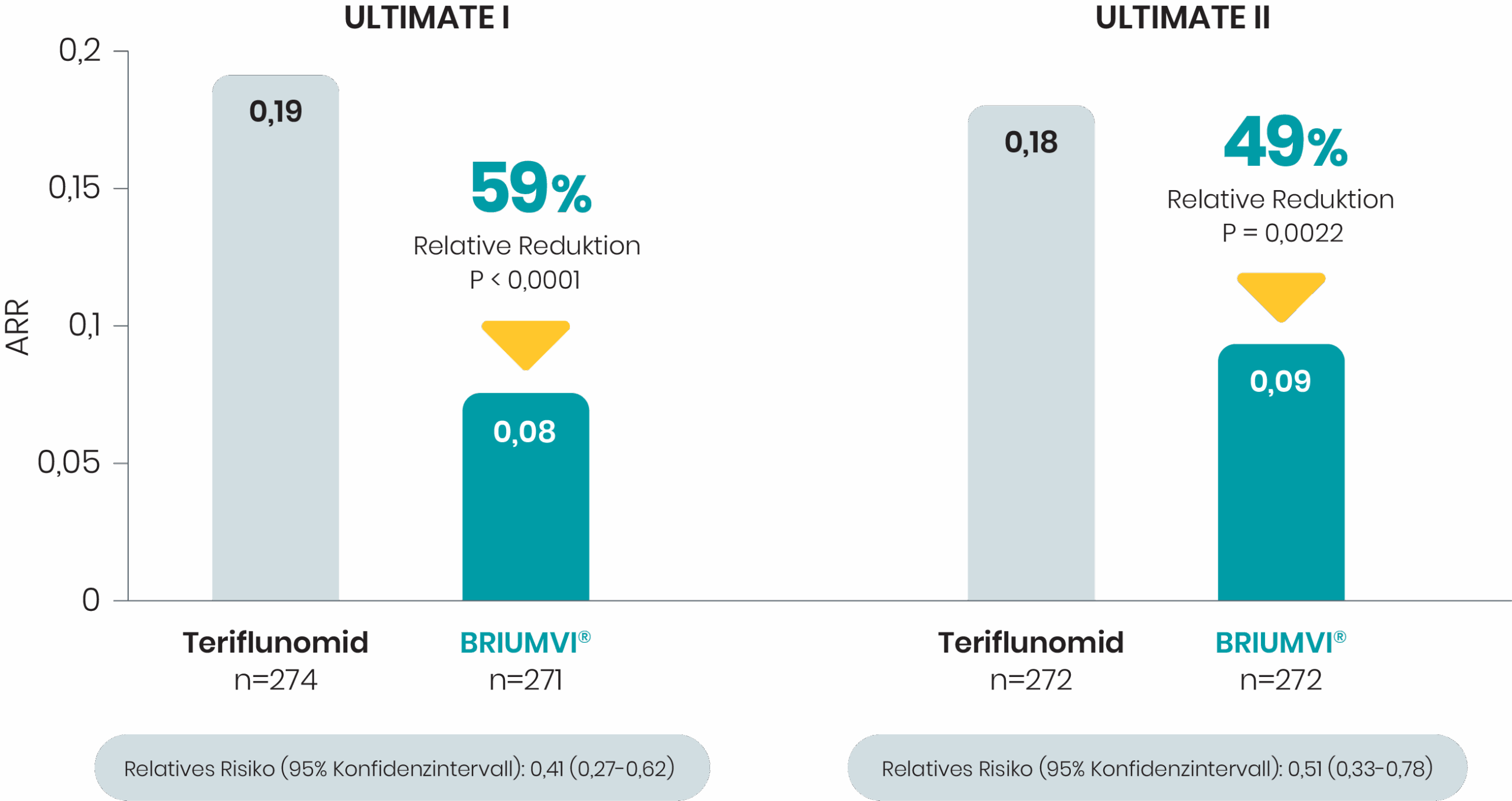
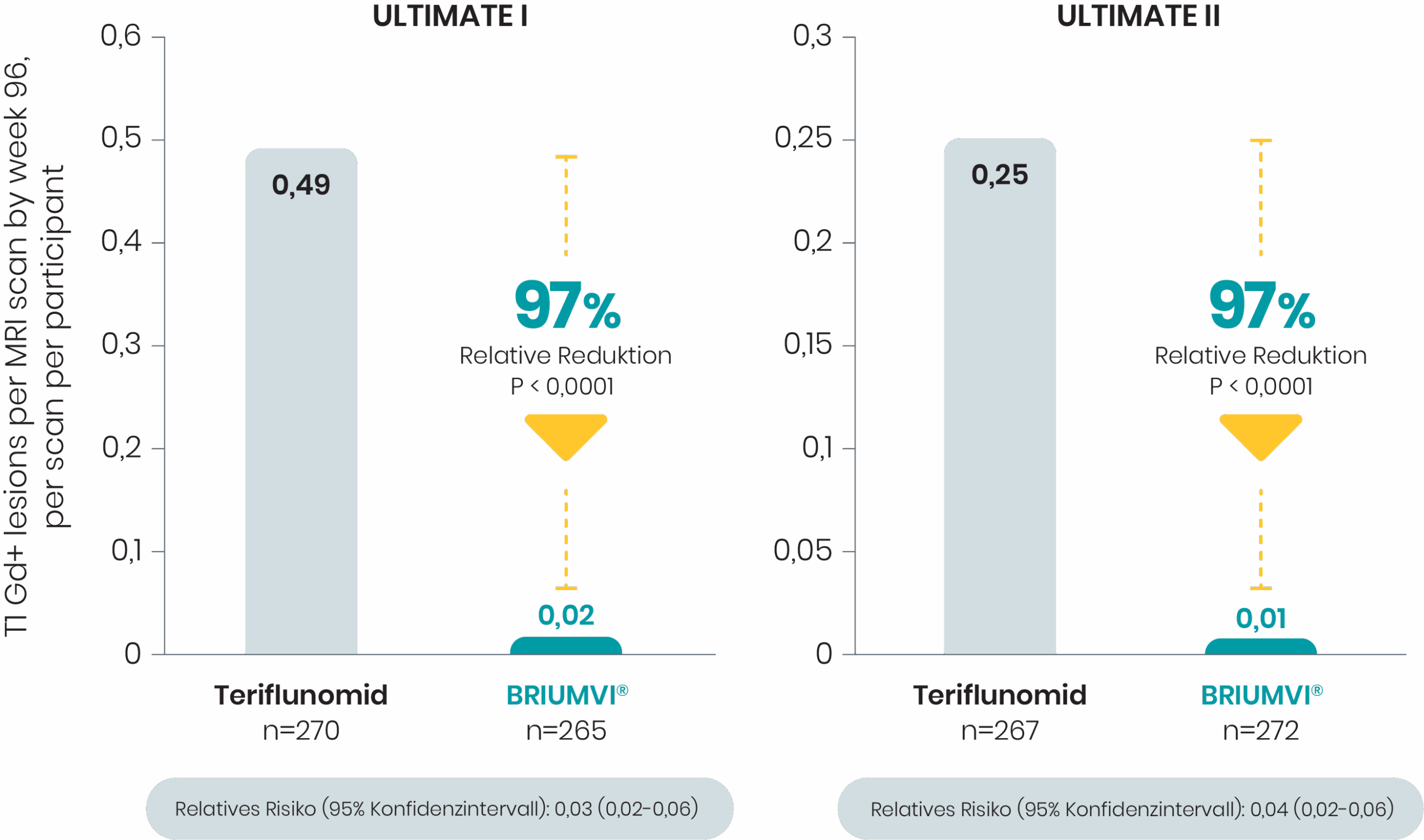
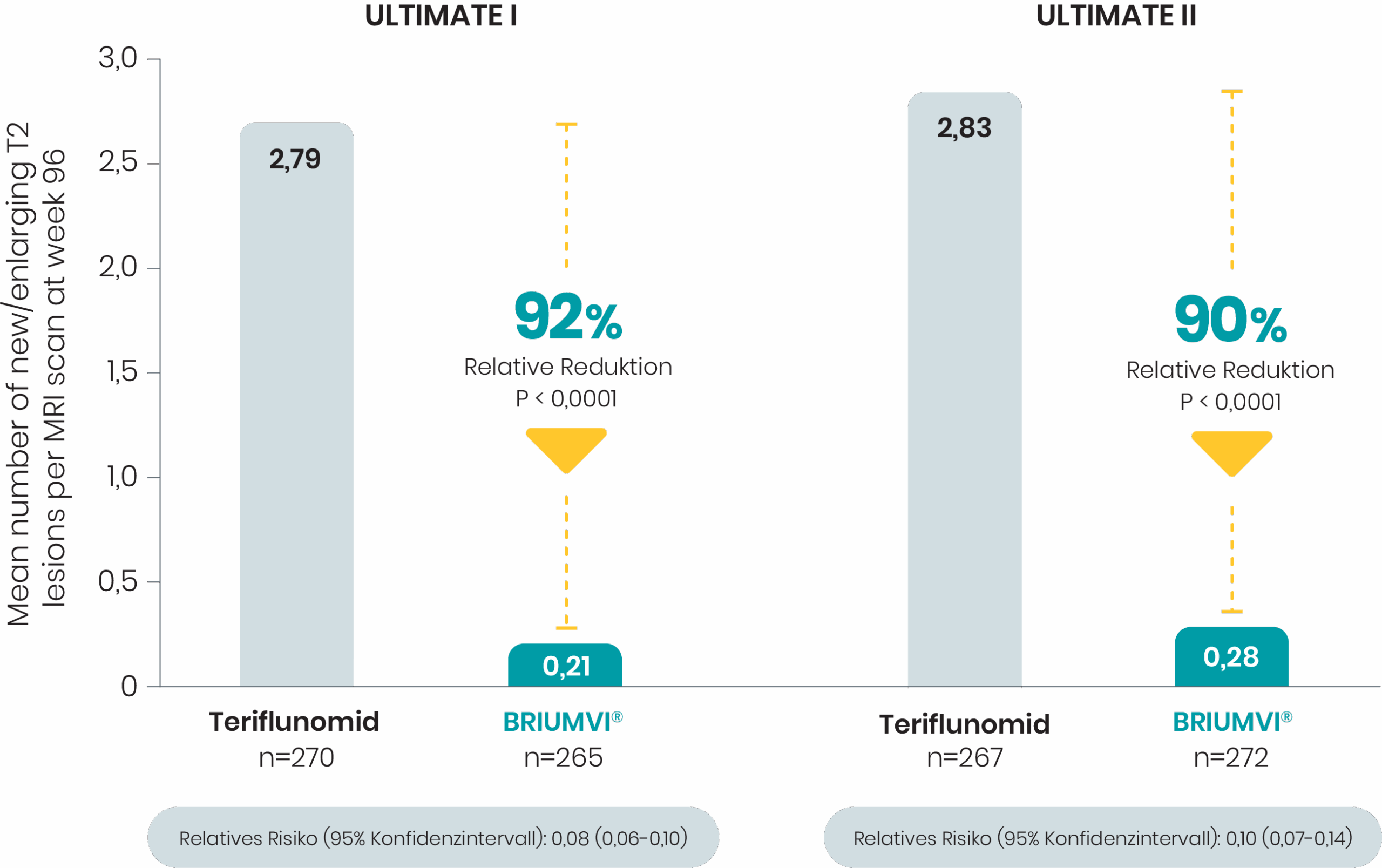
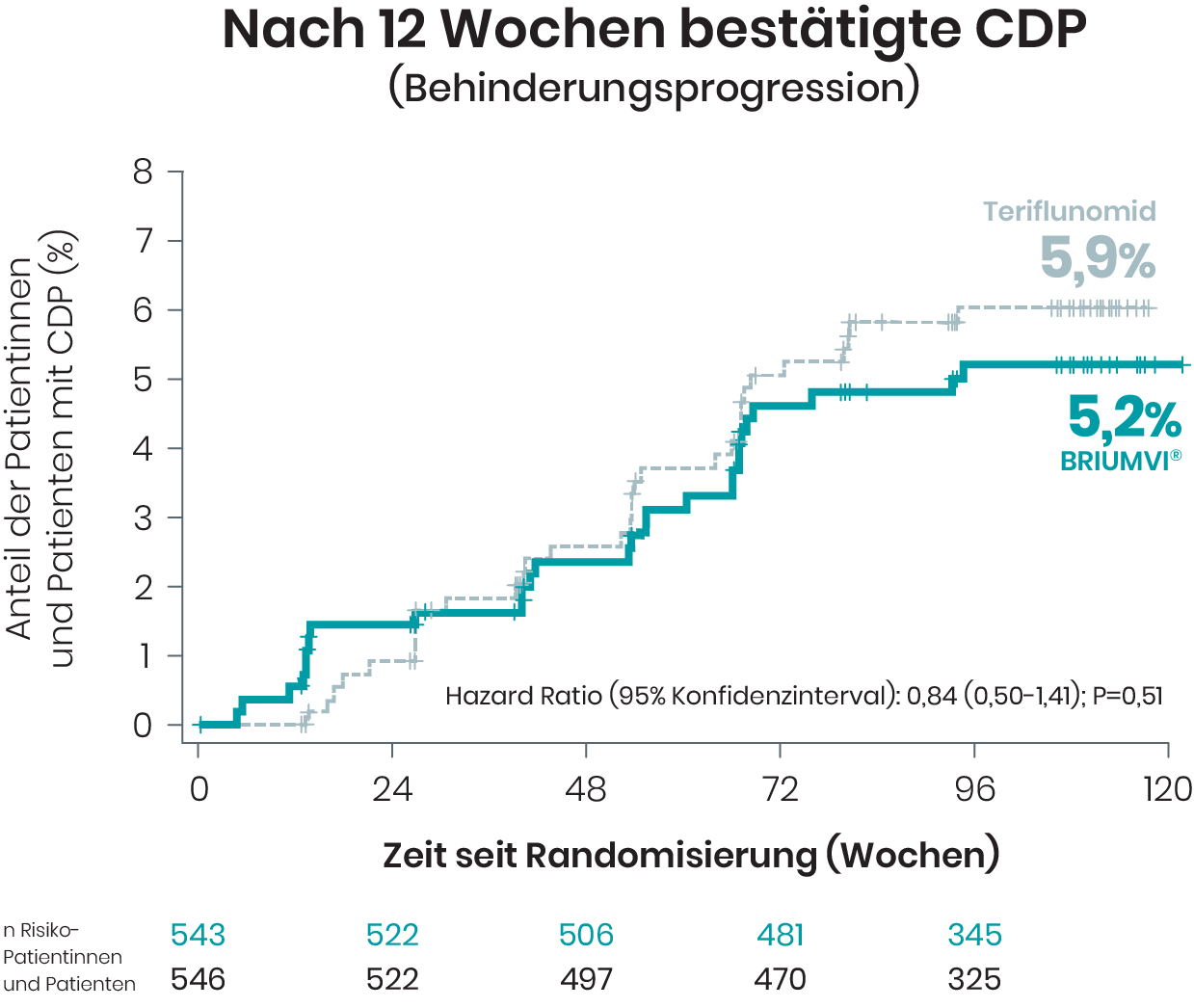
2Steinman L, Fox E, Hartung H-P, et al. Ublituximab versus Teriflunomid bei schubförmiger Multipler Sklerose. N Engl J Med. 2022;387(8):704-714. doi:10.1056/NEJMoa2201904.
3Wattjes MP, Ciccarelli O, Reich DS, et al. 2021 MAGNIMS-CMSC-NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. 2021;20(8):653-670. doi:10.1016/S1474-4422(21)00095-8.
4Hauser SL, Bar-Or A, Comi C, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017; 376(3): 221-234. doi: 10.1056/NEJMoa1601277.
5Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis N Engl J Med. 2020; 383(6): 546-557. doi: 10.1056/NEJMoa1917246.
6Mitsikostas DD, and Goodin DS. Comparing the efficacy of disease-modifying therapies in multiple sclerosis. Mult Scler Relat Disord. 2017; 18: 109-116. doi: 10.1016/j.msard.2017.08.003..
7Alvarez E, Steinman L, Fox EJ, Hartung H-P, Qian P, Wray S, Robertson D, Selmaj K, Wynn D, Mok K, Xu Y, Bodhinathan K, Miskin HP and Cree BAC (2024) Improvements in no evidence of disease activity with ublituximab vs.
teriflunomide in the ULTIMATE phase 3 studies in relapsing multiple sclerosis. Front. Neurol. 15:1473284. doi: 10.3389/fneur.2024.1473284.
8Giovannoni G, Turner B, Gnanapavan S, Offiah C, Schmierer K, Marta M. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult Scler Relat Disord. 2015;4(4):329-333. doi:10.1016/j.msard.2015.04.006.